To obtain a continuous time formulation, we use cubic B-splines, which are sets of piecewise defined cubic polynomials, to represent temporal gene expression profiles. Splines in general, and cubic B-splines in particular, are mathematically convenient for data approximation are often used to produce smooth low-degree polynomial curves, while avoiding the problems of overfitting, numerical instability and oscillations that arise if single high-degree polynomials are used.
B-splines are described as a linear combination of a set of basis polynomials. By knowing the value of these splines at a set of control points, one can generate the entire set of polynomials from these basis functions. We assume that the expression of gene ![]() at time
at time ![]() can be represented by a spline curve and additional noise using the following equation:
can be represented by a spline curve and additional noise using the following equation:
Here ![]() is the expression profile for gene
is the expression profile for gene ![]() ,
, ![]() is a vector of spline control points for gene
is a vector of spline control points for gene ![]() and
and ![]() is a matrix of spline basis functions evaluated at the sampling points of the experiment.
is a matrix of spline basis functions evaluated at the sampling points of the experiment. ![]() is a vector of the noise terms, which is assumed to be normally distributed with mean
is a vector of the noise terms, which is assumed to be normally distributed with mean ![]() .
Because the data is expected to be noisy, and may contain missing values, determining the parameters of the above equation (
.
Because the data is expected to be noisy, and may contain missing values, determining the parameters of the above equation (![]() and
and ![]() ) for each gene separately may lead to overfitting. Instead, when estimating these splines from expression data, we constrain the control point values of genes in the same cluster (co-expressed genes) to co-vary, thus using other co-expressed genes to overcome noise and missing values for a single gene. The parameters of this model are determined using an EM algorithm. In the E step we determine cluster membership for each gene, while the other parameters of the model are maximized with respect to cluster assignment in the M step. See [#!BGGJS03!#] for complete details.
) for each gene separately may lead to overfitting. Instead, when estimating these splines from expression data, we constrain the control point values of genes in the same cluster (co-expressed genes) to co-vary, thus using other co-expressed genes to overcome noise and missing values for a single gene. The parameters of this model are determined using an EM algorithm. In the E step we determine cluster membership for each gene, while the other parameters of the model are maximized with respect to cluster assignment in the M step. See [#!BGGJS03!#] for complete details.
Given the spline coefficient matrix, ![]() , in the above model, for a data set containing
, in the above model, for a data set containing ![]() time-points when
time-points when ![]() control points are used, the smoothing matrix
control points are used, the smoothing matrix ![]() is defined as:
is defined as:
| (2) |
where ![]() is a
is a ![]() matrix. Thus rows of
matrix. Thus rows of ![]() define the ``coordinates'' of the time-points in terms of the basis splines.
define the ``coordinates'' of the time-points in terms of the basis splines.
In previous work [Bar-Joseph et al. 2002], we showed that this method provides a superior fit for time series expression data when compared to all other previous methods.
|
![\includegraphics[width=3.4in]{methods1_t8_fig1.eps}](img14.png)
![\includegraphics[width=1.8in]{methods2_t8_fig2.eps}](img15.png)
![\includegraphics[width=4.5in]{sim_dAEI_ai.eps}](img19.png)
|
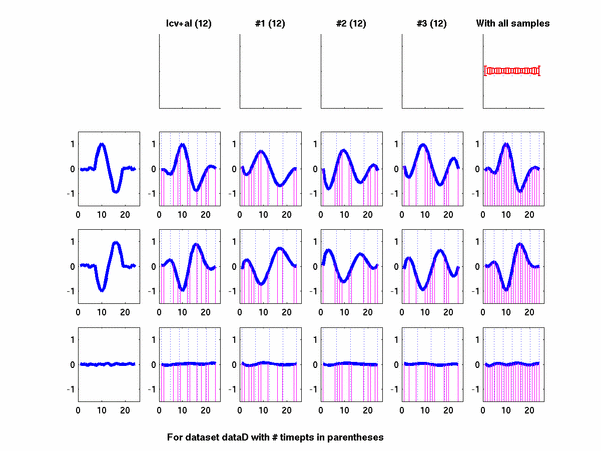
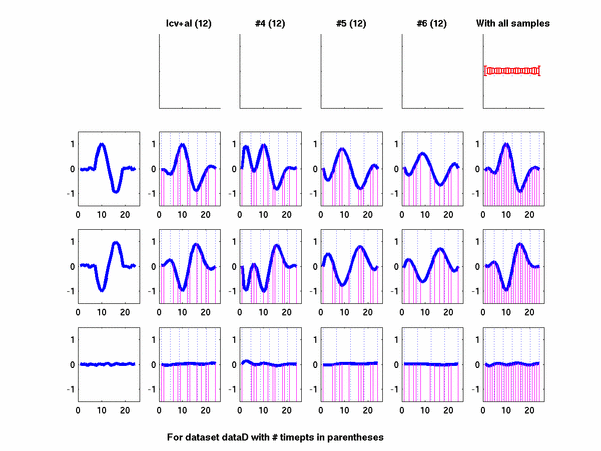
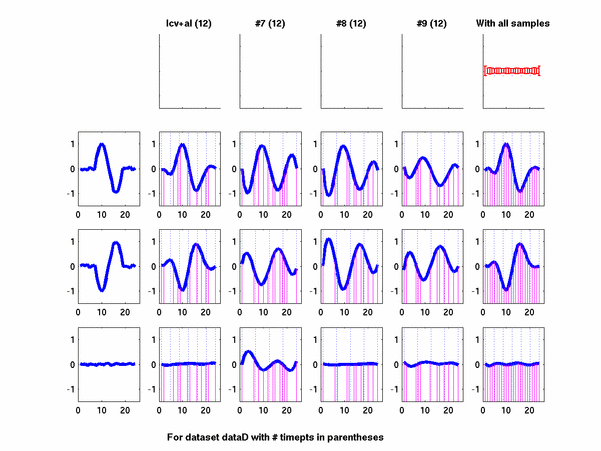
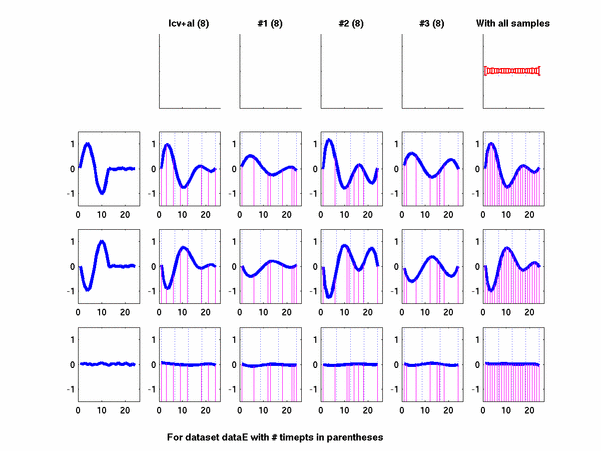
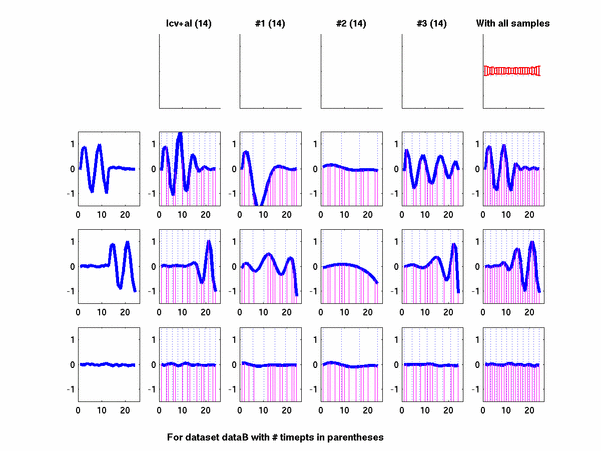
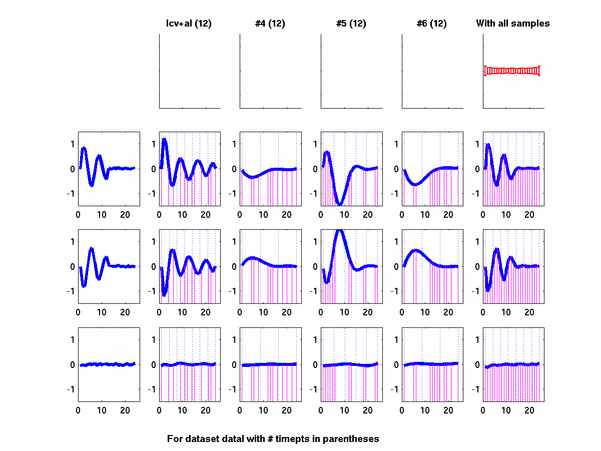
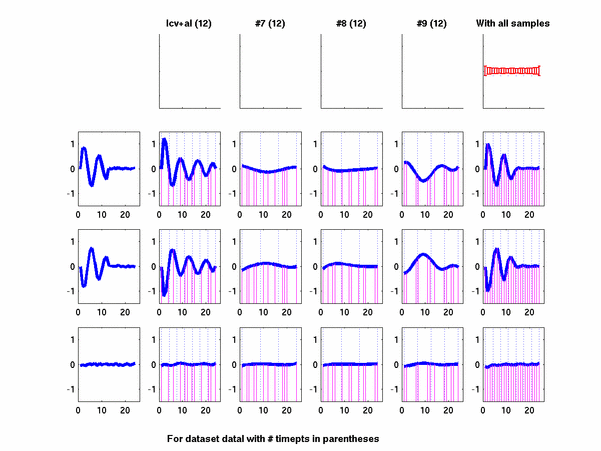
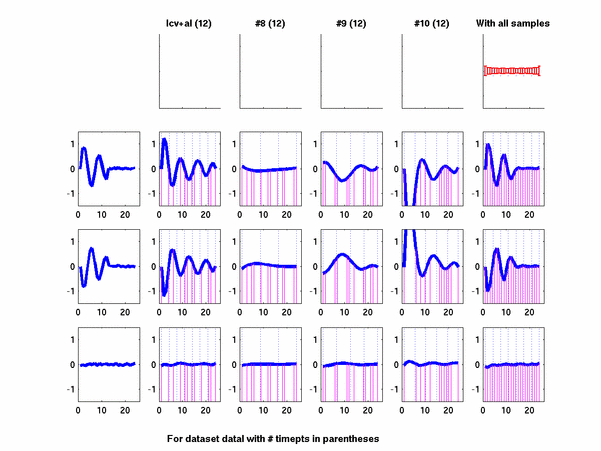
The hardness of a dataset is controlled by varying the frequency of sinusoids and their positions across the clusters: higher frequency implies that more time-points will be needed to characterize the curve in that region. Also, introducing flat regions makes the dataset harder in the opposite way: if we require that sampling strategy use fewer time-points in total, then it will then have to find out a way to identify flat regions and sample at a low rate there.
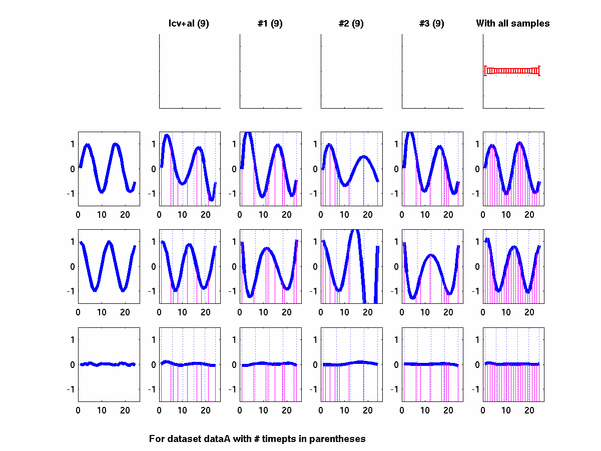
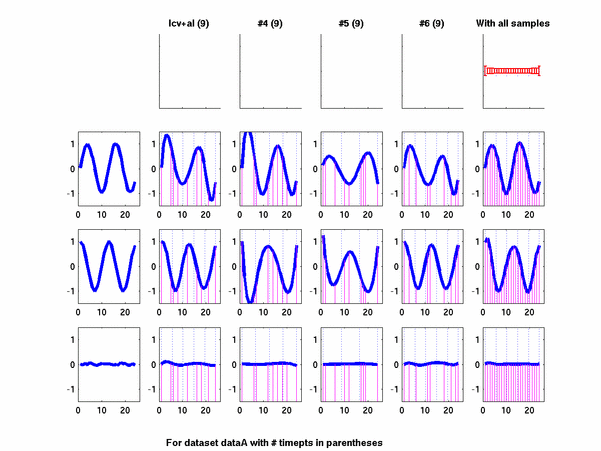
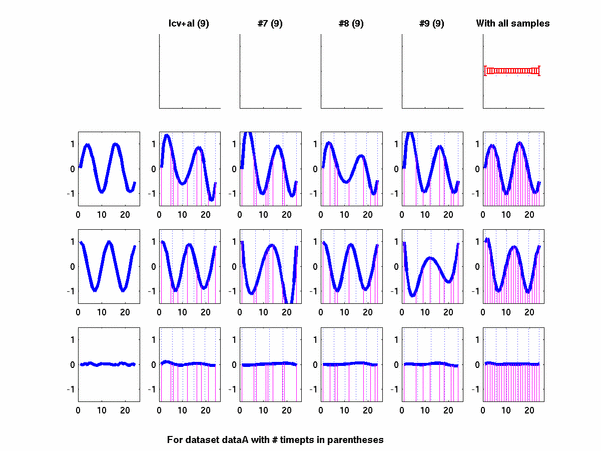
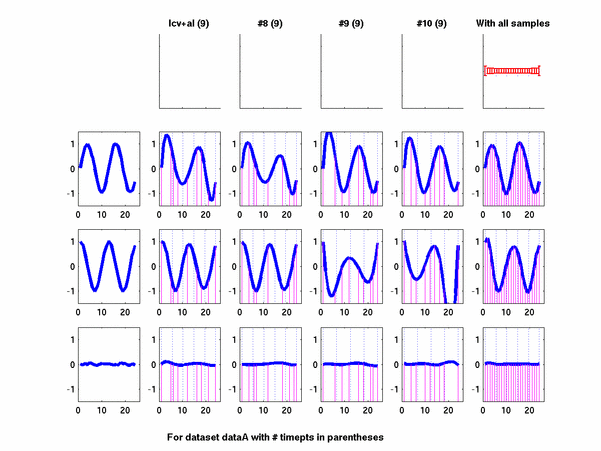
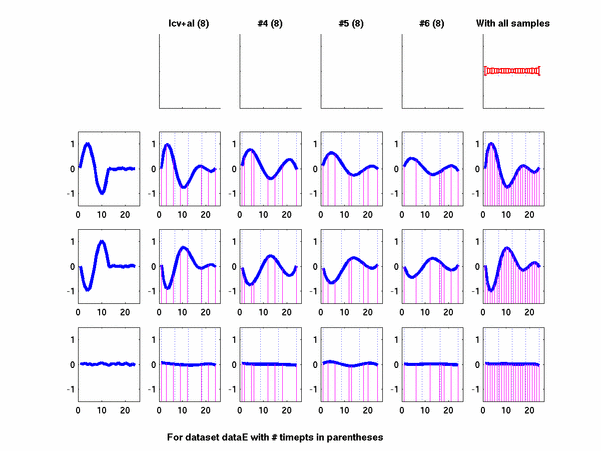
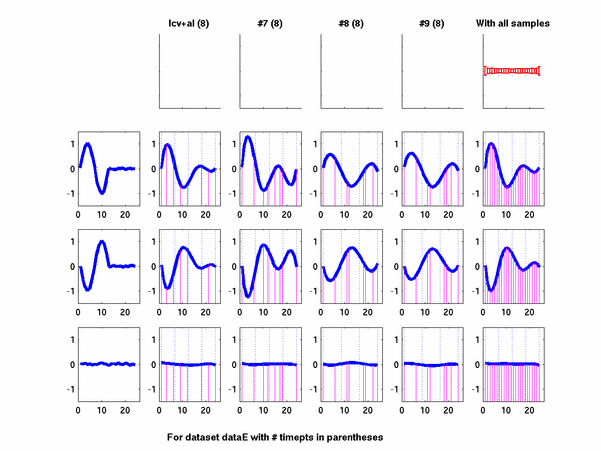
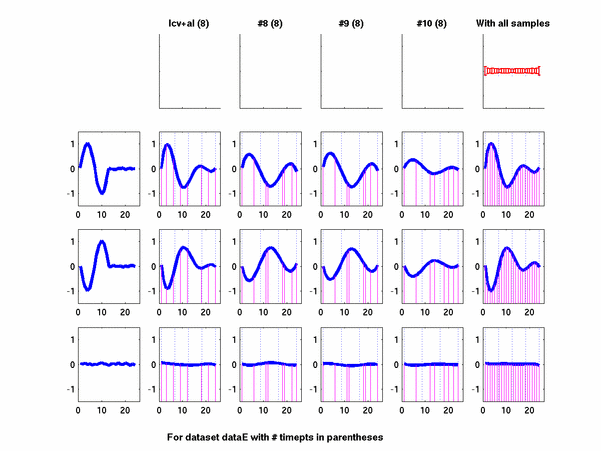
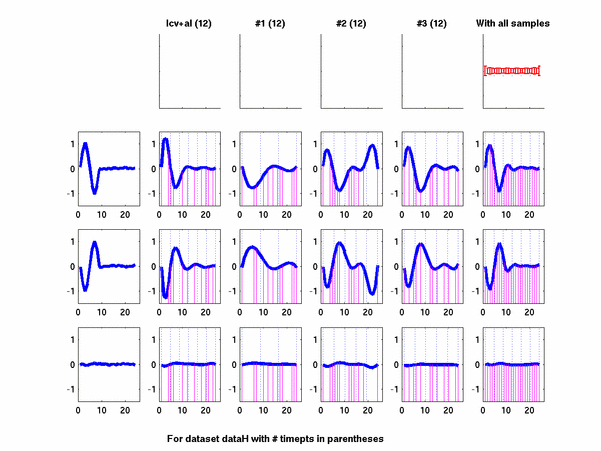
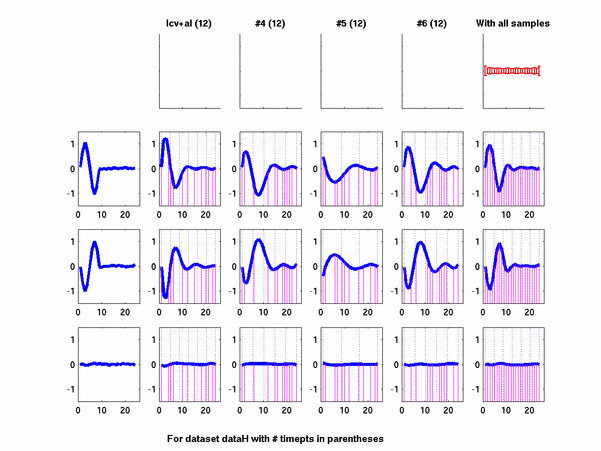
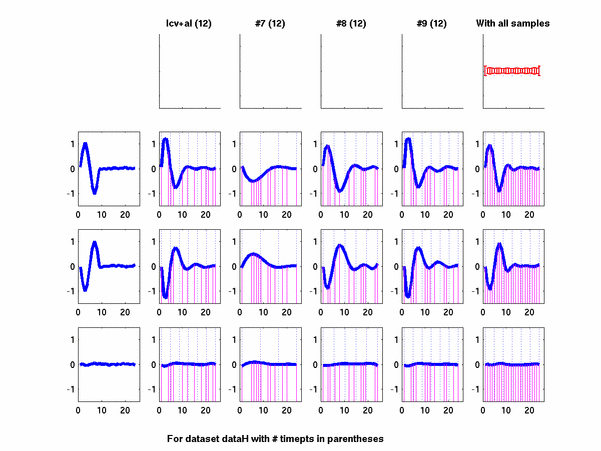
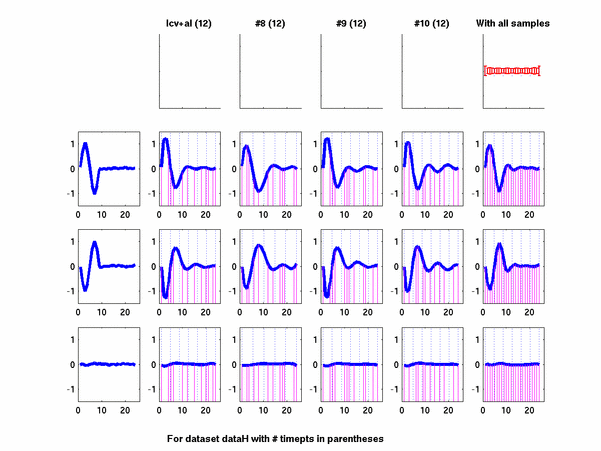
Plots for some other generated datasets are shown here:
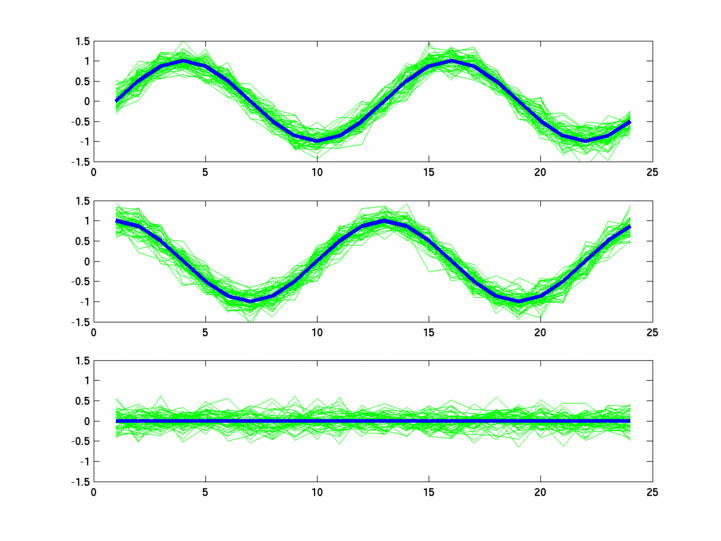{kind=link}
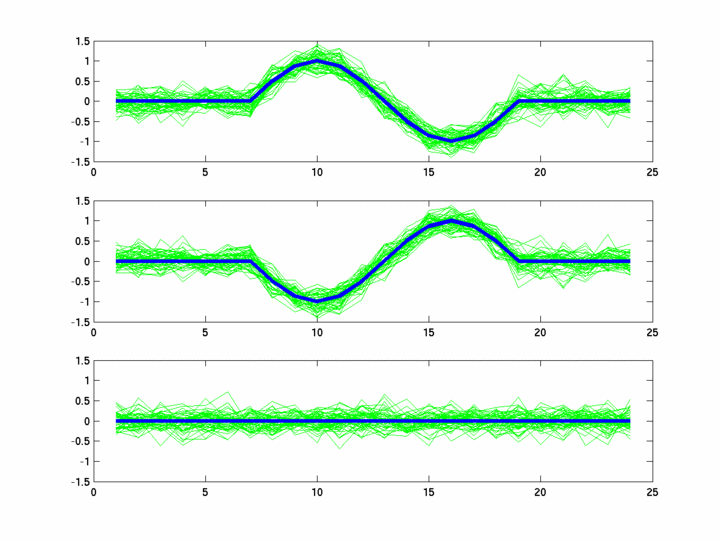{kind=link}
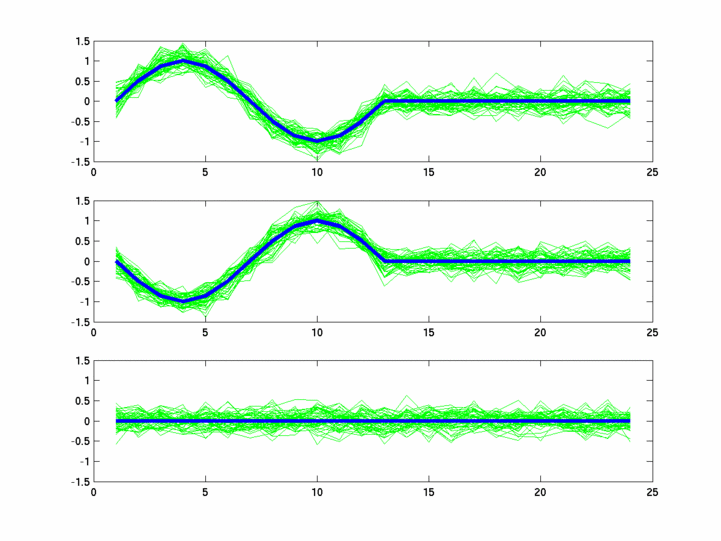{kind=link}
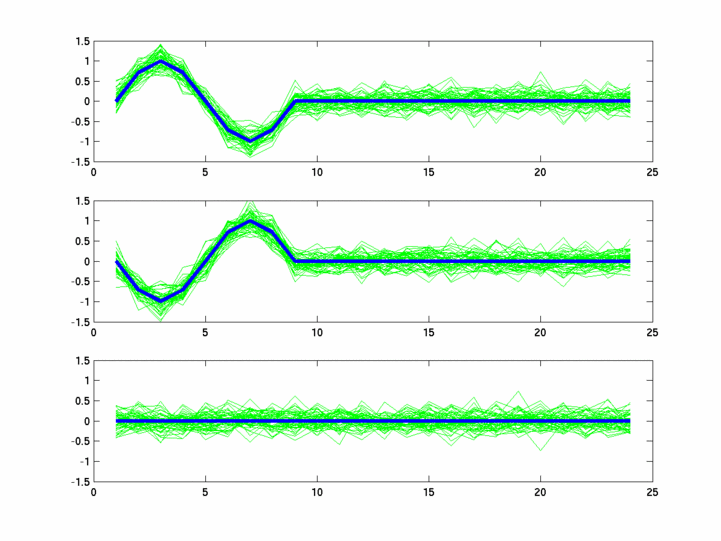{kind=link}
{kind=link}
{kind=link}
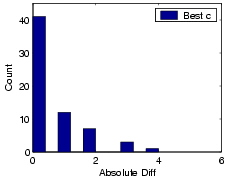
The adaptive cost strategy is quite intuitive: if the addition of new data
doesn't change ![]() , decrease the cost a bit; otherwise, set it back to the
default value (= 1). More precisely,
, decrease the cost a bit; otherwise, set it back to the
default value (= 1). More precisely,
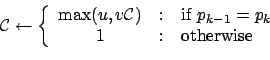
where
- Easy:
- Moderate:
- Hard:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
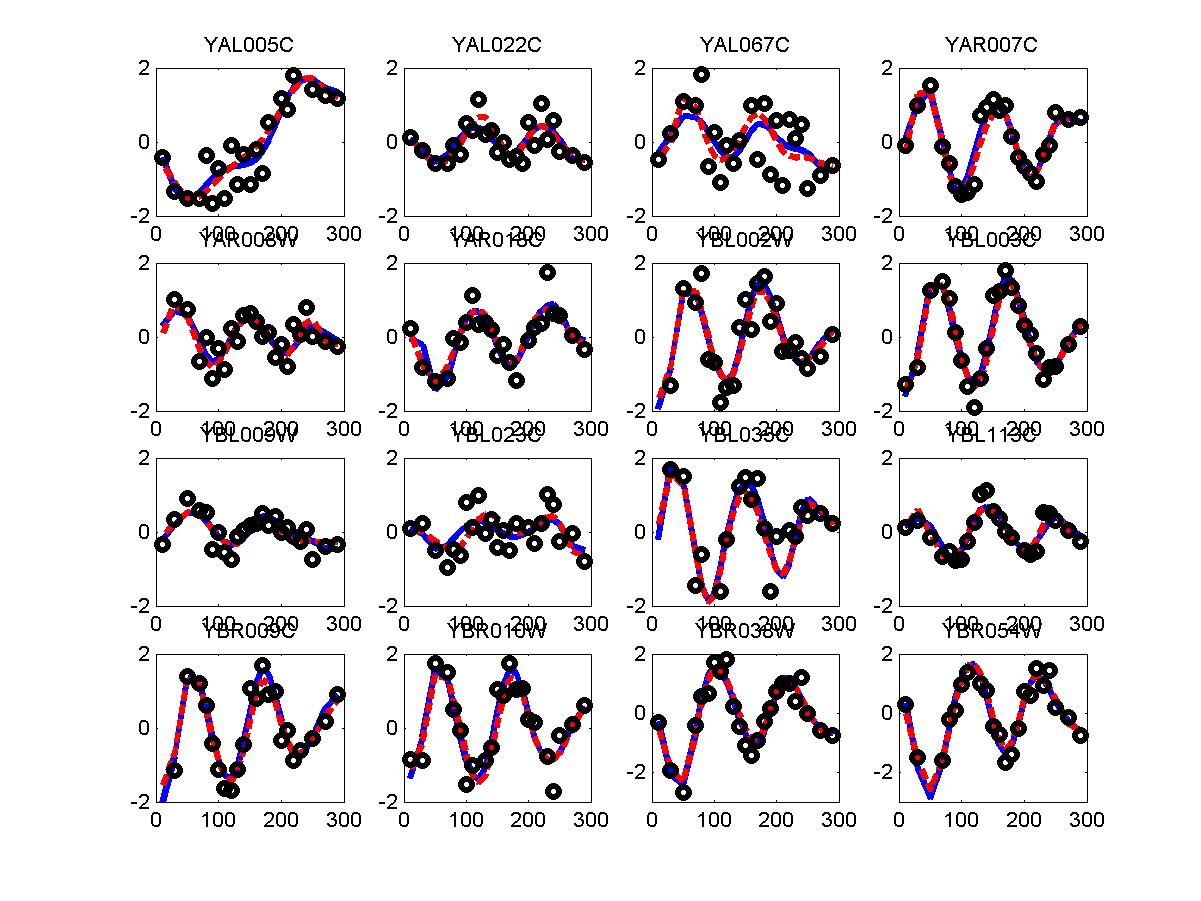
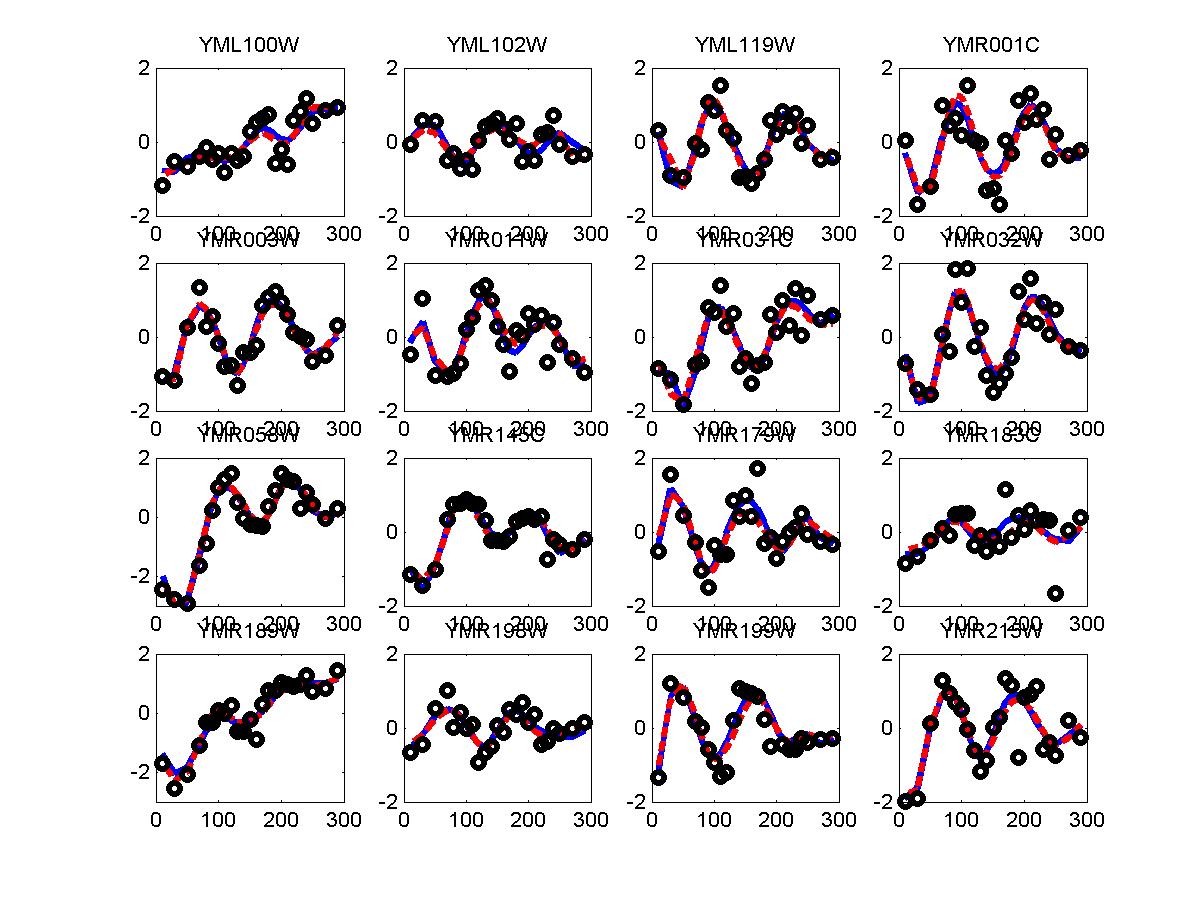
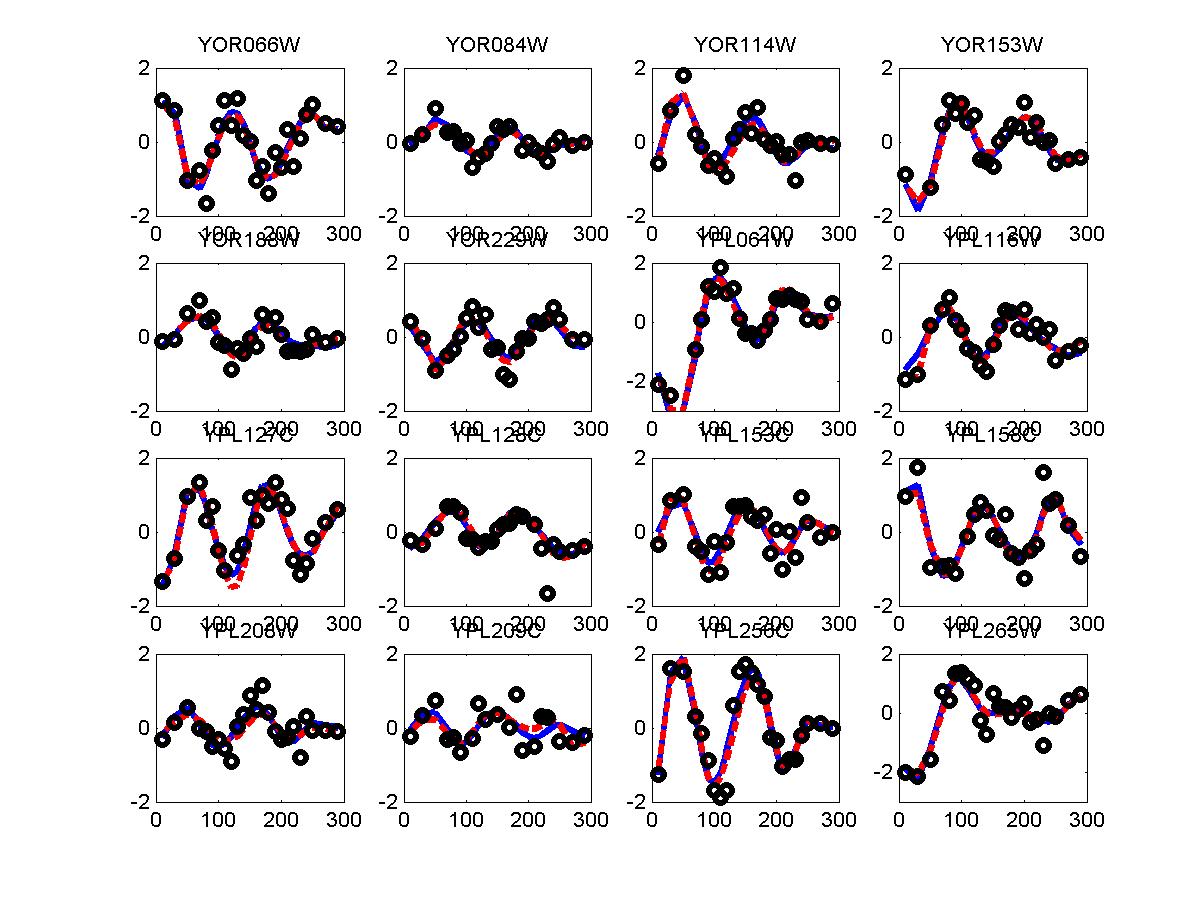
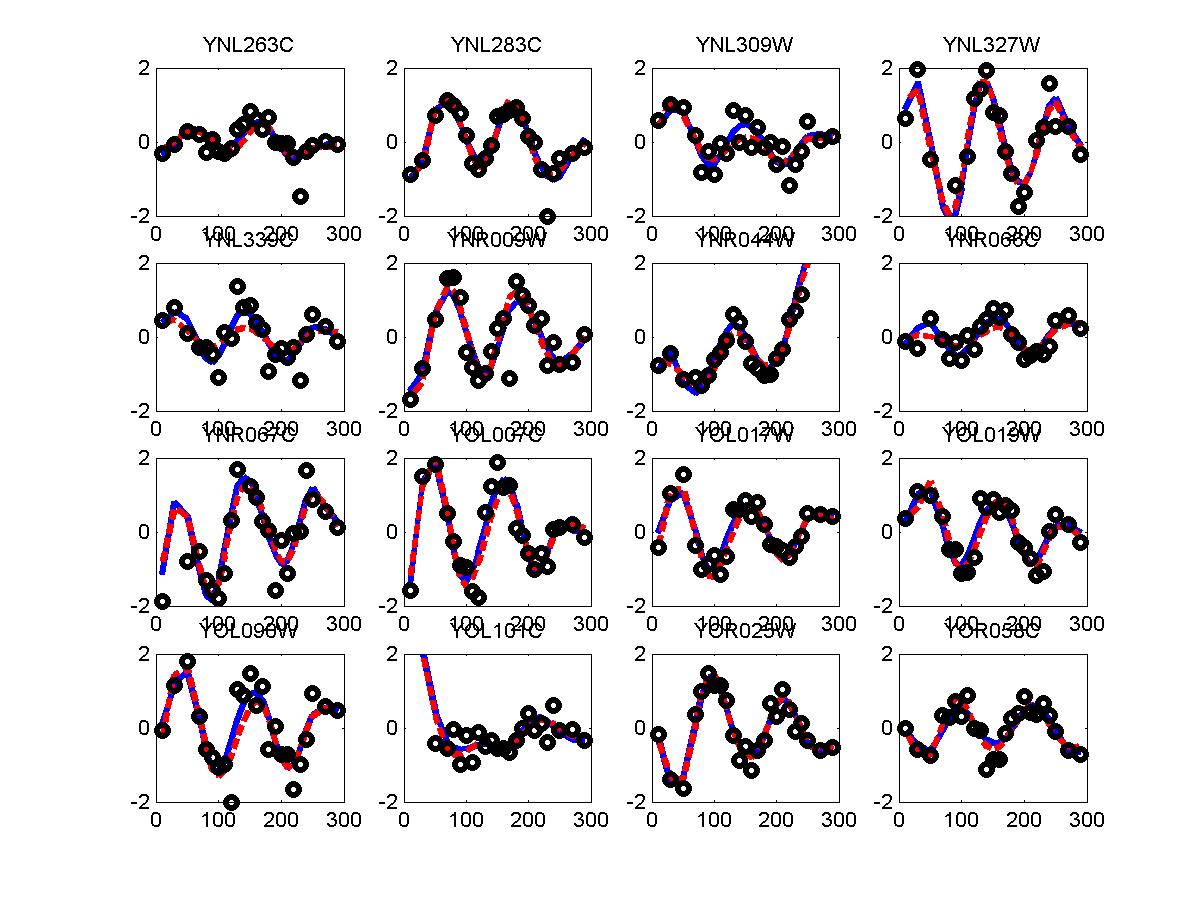
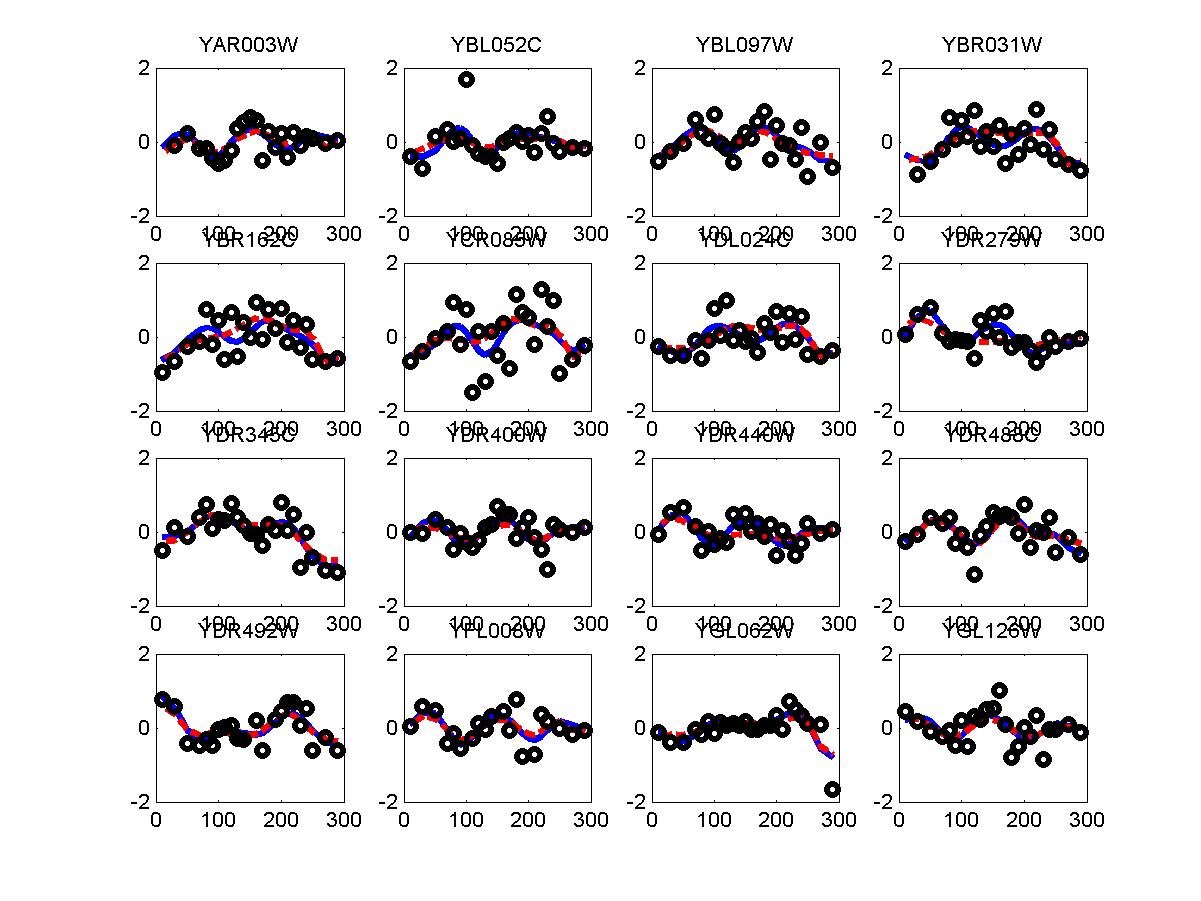
We have used the periodogram* to rank order all genes in the alpha and cdc28 experiments. Periodogram uses Fourier analysis to determine which genes cycle during the experiment. For both experiments we used the cell cycle duration supplied in the Spellman paper (64 minutes for alpha and 85 minutes for cdc28). FV and FC28 consisted of the top 500 genes according to the ranked lists of alpha and cdc28 respectfully. In order to generate a consensus set from both datasets we summed the rank of the gene in both lists (so that a gene that was ranked 50 in alpha and 34 in cdc28 received a score of 84). We then selected the 500 genes with the lowest score and used these as our consensus set which we denoted in the paper as FCOMB.
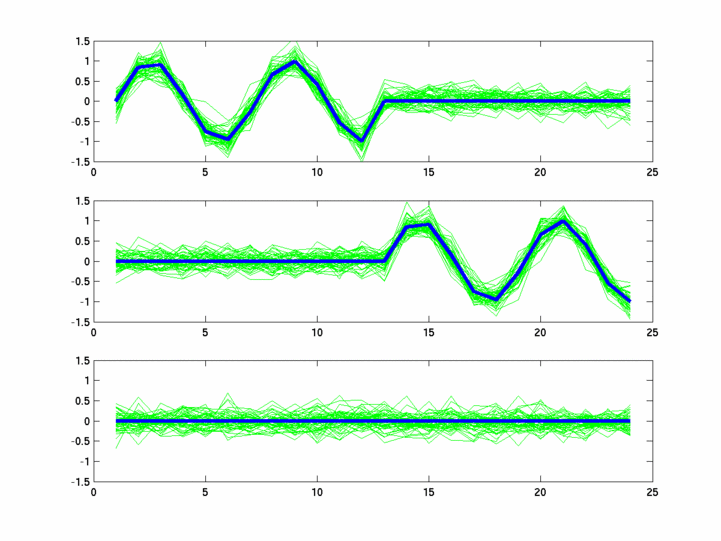
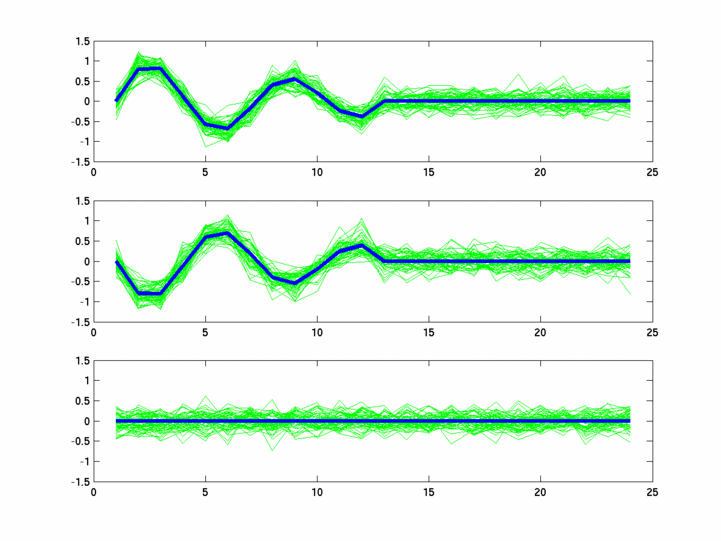
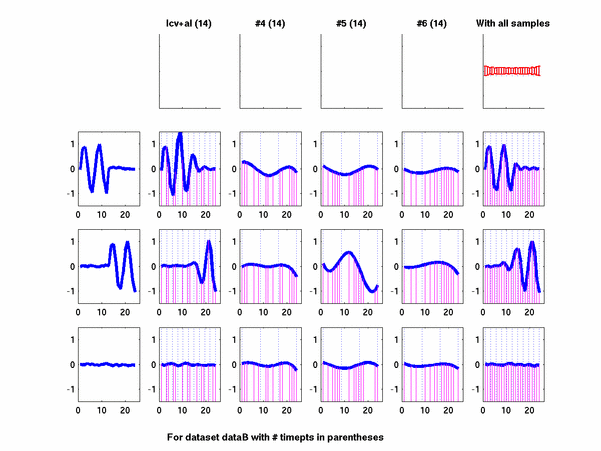
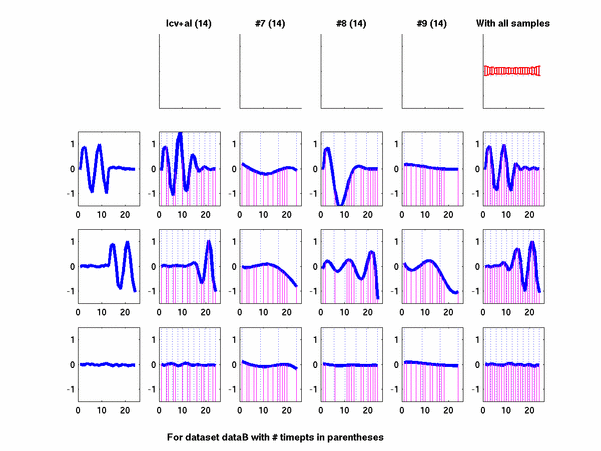
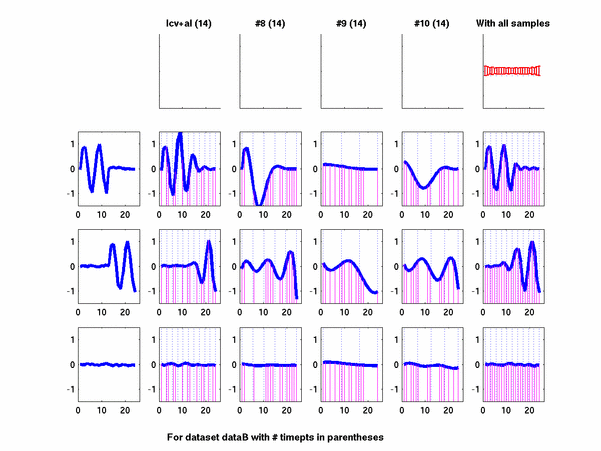
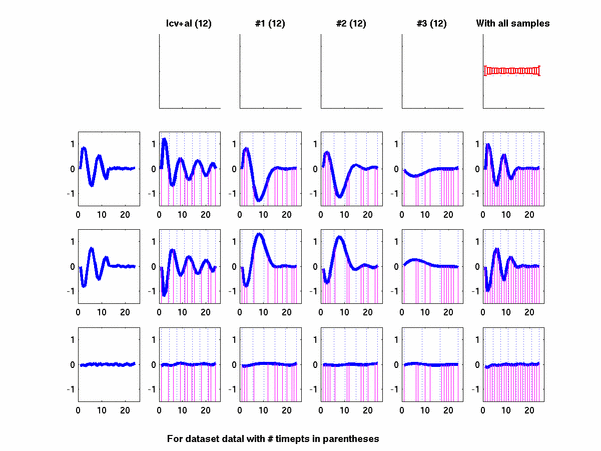
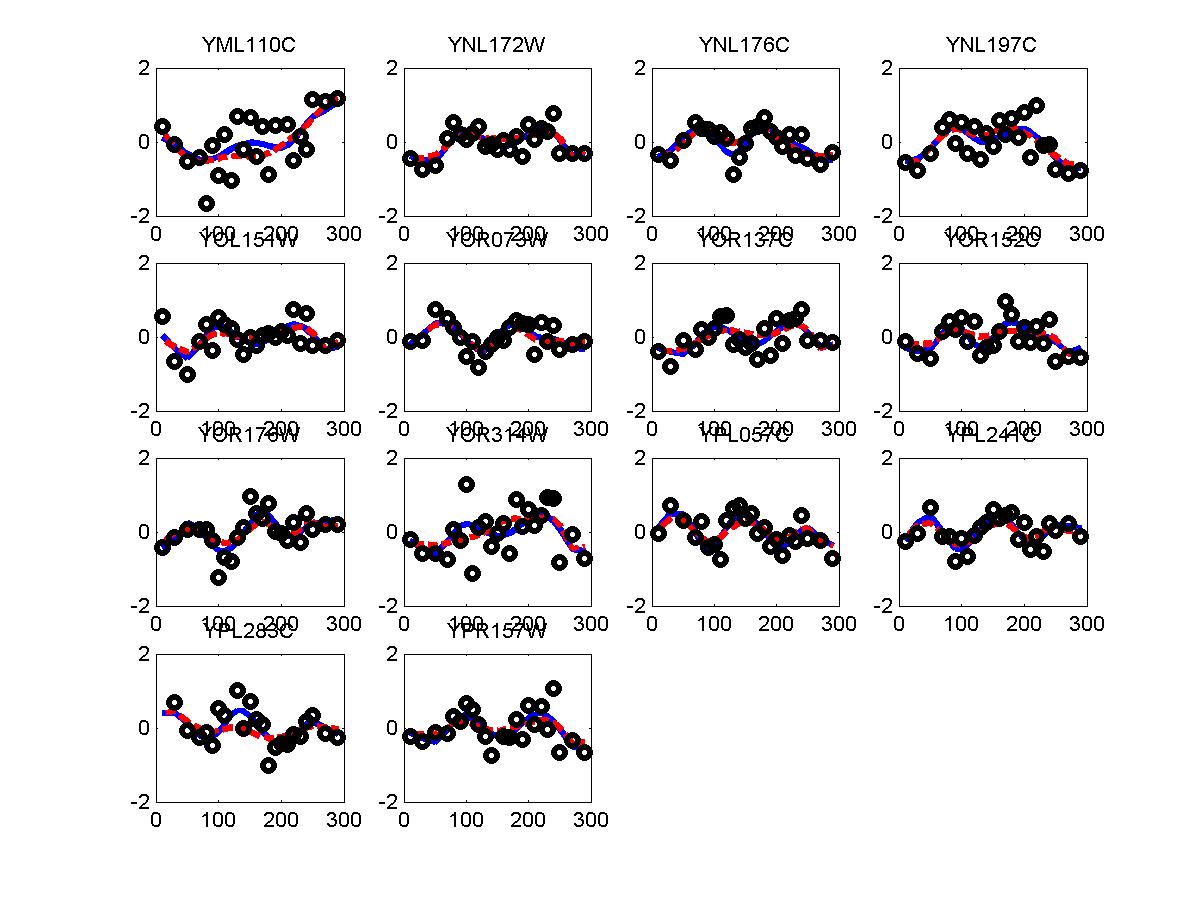
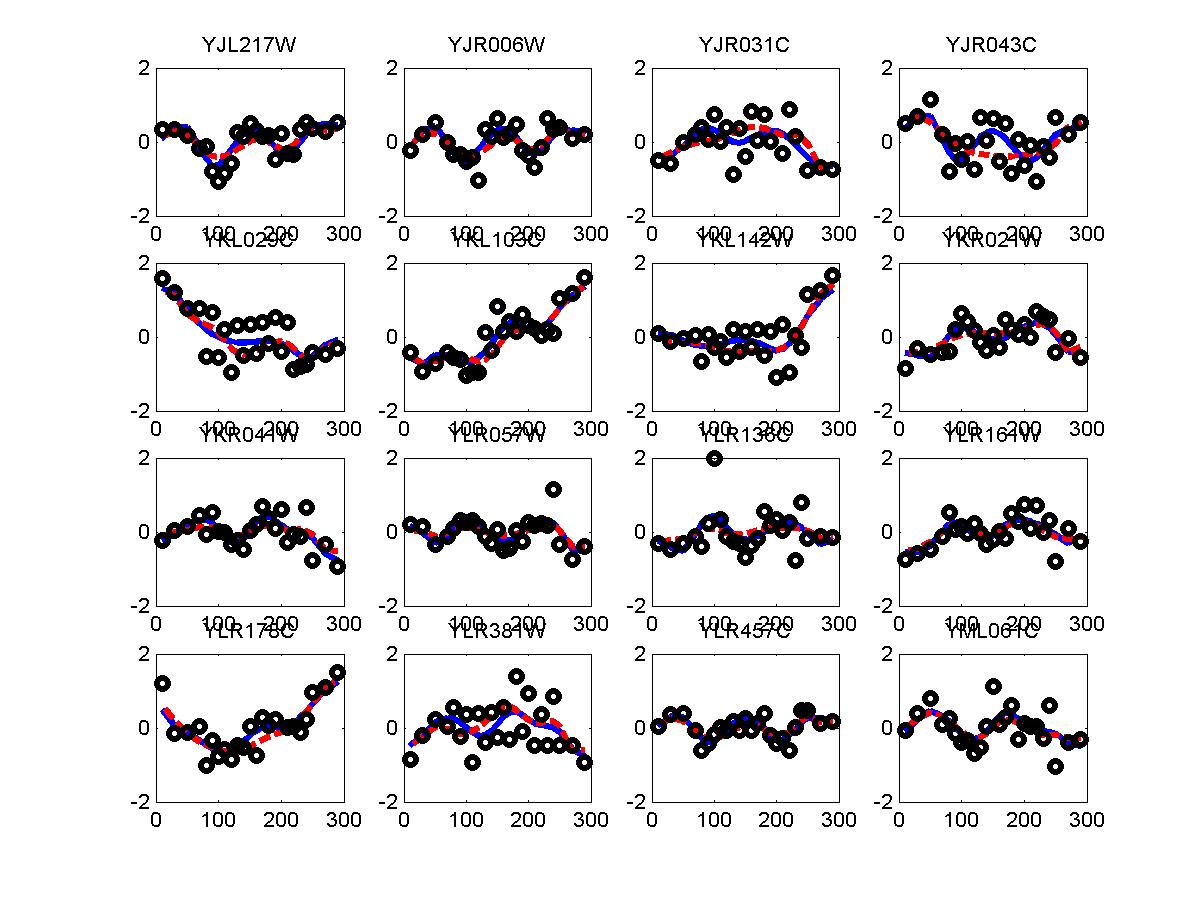
*: See Ref 21 in paper: Wicher, Fokianos and Strimmer, Bioinformatics 20:5-20, 2004.- Example of cycling genes which can be found using only 20 (out of 24) time-points: Fig #1 , Fig #2 , Fig #3 , Fig #4.
- Cycling genes where all 24 time-points are necessary to identify them as cycling: Fig #1, , Fig #2, , Fig #3. Note that even in these genes the recovered expression profile using only 20 time-points, is very similar to the profile recovered using all 24 time-points.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}